
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined silica supported aluminum hydride: another step towards the utopian single site dream?†
Baraa
Werghi
,
Anissa
Bendjeriou-Sedjerari
,
Julien
Sofack-Kreutzer
,
Abdesslem
Jedidi
,
Edy
Abou-Hamad
,
Luigi
Cavallo
* and
Jean-Marie
Basset
*
KAUST Catalysis Center (KCC, ), King Abdullah University of Science and Technology, Thuwal-23955-6900, Saudi Arabia. E-mail: luigi.cavallo@kaust.edu.sa; jeanmarie.basset@kaust.edu.sa
First published on 20th July 2015
Abstract
Reaction of triisobutylaluminum with SBA15700 at room temperature occurs by two parallel pathways involving either silanol or siloxane bridges. It leads to the formation of a well-defined bipodal [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)2Al–CH2CH(CH3)2] 1a, silicon isobutyl [Si–CH2CH(CH3)2] 1b and a silicon hydride [Si–H] 1c. Their structural identity was characterized by FT-IR and advanced solid-state NMR spectroscopies (1H, 13C, 29Si, 27Al and 2D multiple quantum), elemental and gas phase analysis, and DFT calculations. The reaction involves the formation of a highly reactive monopodal intermediate: [SiO–Al–[CH2CH(CH3)2]2], with evolution of isobutane. This intermediate undergoes two parallel routes: transfer of either one isobutyl fragment or of one hydride to an adjacent silicon atom. Both processes occur by opening of a strained siloxane bridge, Si–O–Si but with two different mechanisms, showing that the reality of “single site” catalyst may be an utopia: DFT calculations indicate that isobutyl transfer occurs via a simple metathesis between the Al-isobutyl and O–Si bonds, while hydride transfer occurs via a two steps mechanism, the first one is a β-H elimination to Al with elimination of isobutene, whereas the second is a metathesis step between the formed Al–H bond and a O–Si bond. Thermal treatment of 1a (at 250 °C) under high vacuum (10−5 mbar) generates Al–H through a β-H elimination of isobutyl fragment. These supported well-defined Al–H which are highly stable with time, are tetra, penta and octa coordinated as demonstrated by IR and 27Al–1H J-HMQC NMR spectroscopy. All these observations indicate that surfaces atoms around the site of grafting play a considerable role in the reactivity of a single site system.
SiO)2Al–CH2CH(CH3)2] 1a, silicon isobutyl [Si–CH2CH(CH3)2] 1b and a silicon hydride [Si–H] 1c. Their structural identity was characterized by FT-IR and advanced solid-state NMR spectroscopies (1H, 13C, 29Si, 27Al and 2D multiple quantum), elemental and gas phase analysis, and DFT calculations. The reaction involves the formation of a highly reactive monopodal intermediate: [SiO–Al–[CH2CH(CH3)2]2], with evolution of isobutane. This intermediate undergoes two parallel routes: transfer of either one isobutyl fragment or of one hydride to an adjacent silicon atom. Both processes occur by opening of a strained siloxane bridge, Si–O–Si but with two different mechanisms, showing that the reality of “single site” catalyst may be an utopia: DFT calculations indicate that isobutyl transfer occurs via a simple metathesis between the Al-isobutyl and O–Si bonds, while hydride transfer occurs via a two steps mechanism, the first one is a β-H elimination to Al with elimination of isobutene, whereas the second is a metathesis step between the formed Al–H bond and a O–Si bond. Thermal treatment of 1a (at 250 °C) under high vacuum (10−5 mbar) generates Al–H through a β-H elimination of isobutyl fragment. These supported well-defined Al–H which are highly stable with time, are tetra, penta and octa coordinated as demonstrated by IR and 27Al–1H J-HMQC NMR spectroscopy. All these observations indicate that surfaces atoms around the site of grafting play a considerable role in the reactivity of a single site system.
Introduction
There is a tendency to rationalize the understanding of heterogeneous catalysis by making well-defined model compounds.1–8 The study of the reactivity of these models allows the identification of some elementary steps of heterogeneous catalysis.9–14 One of the simplest ways to make such models compounds is to react the surface silanols (Si–OH) of partially dehydroxylated silica with a metal-alkyl to obtain in principle well defined grafted complexes.2,9,15,16 However, it appeared progressively that depending on the degree of dehydroxylation of a support, mono, bi and multipodal species can be generated.2,9,17–19
In fact there is growing evidence that the grafted complexes interact with the surrounding atoms present on the surface (Scheme 1).2,9,20
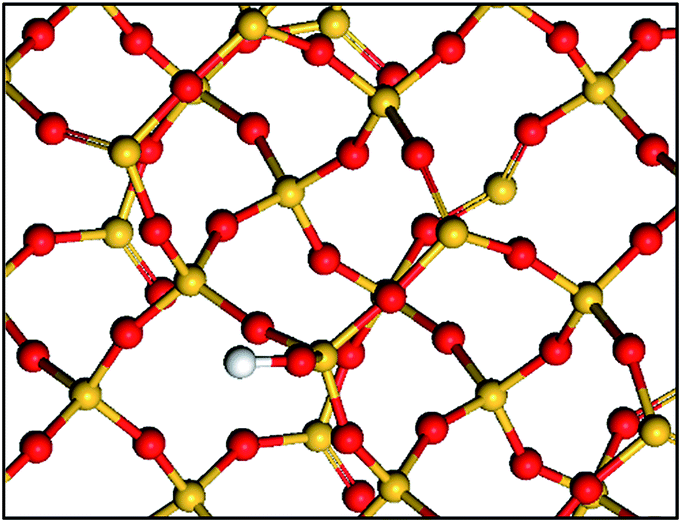 | ||
| Scheme 1 Simple representation of a partially dehydroxylated silica surface: isolated silanols and siloxane bridges. Si atoms are in yellow, O atoms are in red and H atoms are in white. | ||
Indeed, the silica dehydroxylation treatment leads to a highly strained reactive surface that may participate in anchoring reactions.21–25 The resulting surface organometallic chemistry became more associated to an “ensemble” of surfaces atoms that makes this chemistry increasingly difficult to fully understand, approaching to some extent the complexity of classical heterogeneous catalysis (the concept of ensemble effect was invented in heterogeneous catalysis by metals).26–28
We present here an exemplary case of this complexity by studying a well-known organometallic compound which presents the advantage of being monomeric in solution and which has a highly oxophilic metallic atom (Al) susceptible of making strong bond(s) with the silica surface. We demonstrate how the well-defined surface precursor can be transformed on the silica surface into a supported Al hydride via a mechanism showing the complexity of strained siloxane bridges.
The type of reaction between alkyl aluminum compounds (AlR3, R = Me,29–34 Et,35 iBu…) and silica surface still remains not elucidated despite its huge applications as a co-catalyst for olefins polymerization.36–39 Because many aluminum alkyls exist in solution as dimers or trimers, we have chosen triisobutylaluminum (TIBA) because it is a monomer,40 with the hope it will remain monomeric once it is grafted to the surface. A preliminary account of this chemistry was started on mesoporous silica SBA15700.41 At that time diethyl ether was chosen to stabilize the Al precursor because we wanted to avoid chemical interaction with the surrounding sites. Subsequently, several studies appeared on this subject with various aluminum alkyls grafted on SBA15500 and revealed a more complex behavior.42 This previous study involved the reaction of SBA15 surface, dehydroxylated at moderate temperature (500 °C) with an excess of TIBA (3 eq./SiOH). Applying their experimental conditions, Coperet et al. observed significant incorporation of Al in the bulk of SBA15500. Based on the available data, the authors concluded that, under their experimental conditions, there is remarkable incorporation of Al into the framework of SBA15.35,42
Facing this longstanding complex problem of the reactivity of TiBA with silica, we decided to re-investigate this reaction by a carefully chosen combination of the oxide support and experimental conditions. Here, a simple modification of the experimental conditions as compared to Coperet et al. works42 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between TIBA and SiOH and not 3:1) enables us to describe the stepwise interaction of TIBA with SBA15700 at room temperature avoiding thus the possible incorporation of Al in the bulk of SBA15. It occurs either by reaction with silanols (Scheme 2, pathway a) or by reaction with siloxane bridges Si–O–Si by two parallel pathways (Scheme 2, pathway b). Those materials were used as precursors of Al hydrides: a thermal treatment under vacuum of these grafted Al isobutyl leads, in the absence of hydrogen, to very stable Al hydrides that are tetra, penta or octahedral. Al hydrides43 have been known for almost a century as catalysts for the oligomerization of ethylene to linear α-olefins, and aluminum alkyls are used as cocatalysts in Ziegler–Natta polymerization of olefins.44–50
:1 ratio between TIBA and SiOH and not 3:1) enables us to describe the stepwise interaction of TIBA with SBA15700 at room temperature avoiding thus the possible incorporation of Al in the bulk of SBA15. It occurs either by reaction with silanols (Scheme 2, pathway a) or by reaction with siloxane bridges Si–O–Si by two parallel pathways (Scheme 2, pathway b). Those materials were used as precursors of Al hydrides: a thermal treatment under vacuum of these grafted Al isobutyl leads, in the absence of hydrogen, to very stable Al hydrides that are tetra, penta or octahedral. Al hydrides43 have been known for almost a century as catalysts for the oligomerization of ethylene to linear α-olefins, and aluminum alkyls are used as cocatalysts in Ziegler–Natta polymerization of olefins.44–50
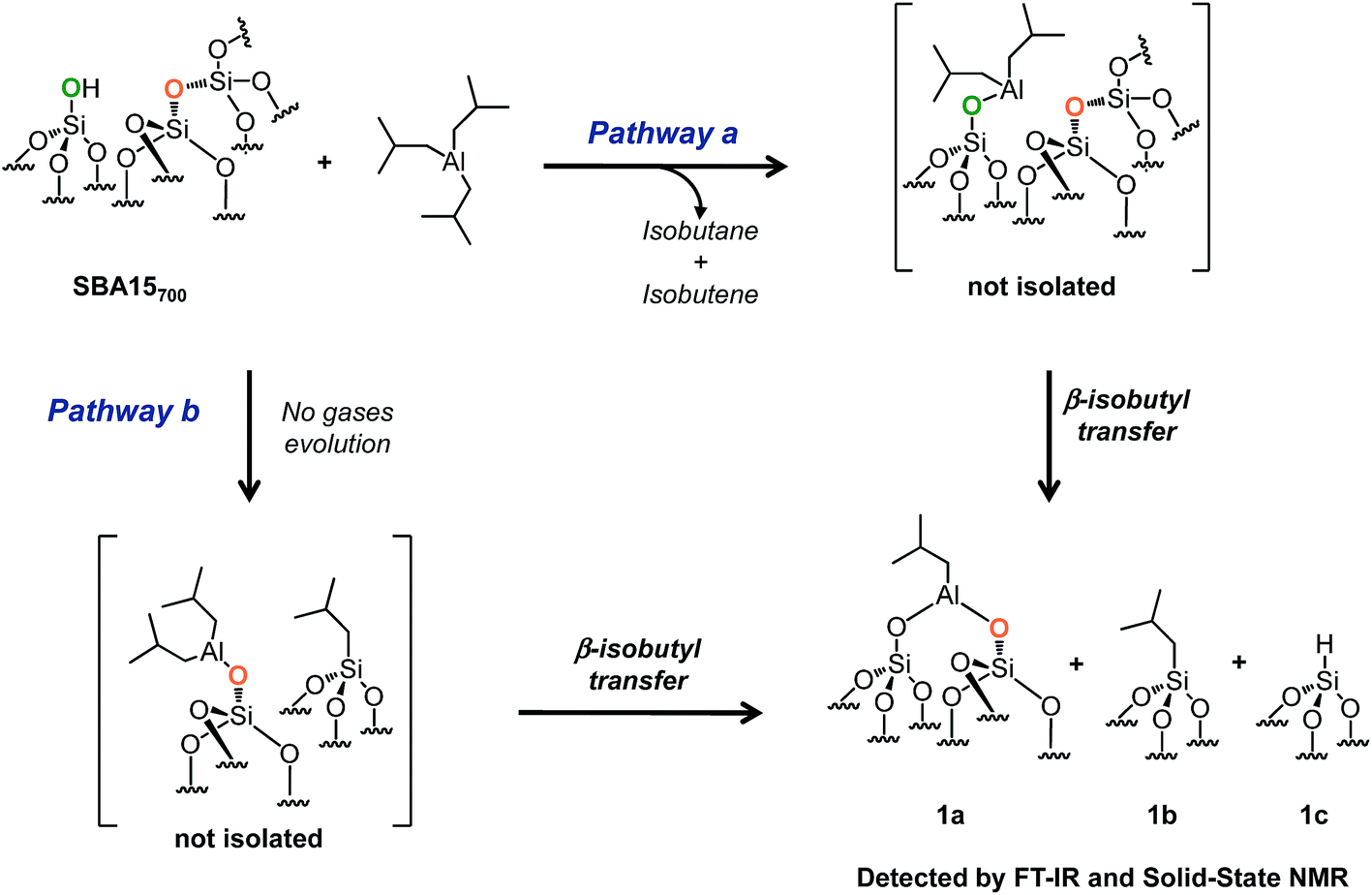 | ||
| Scheme 2 Reaction of TIBA (1 M/hexane) with single silanol (pathway a) and strained siloxane bridges (pathway b) of SBA700 at room temperature. | ||
Results and discussion
Reaction of TIBA with SBA15700 surface
SBA15 is a well-known hexagonal ordered mesoporous support characterized by a high surface area (ESI†), large uniform pore diameter (6 nm), thicker walls (ranging from 3 to 6 nm) and high thermal stability (up to 1200 °C).51,52 After overnight dehydroxylation at 700 °C under high vacuum (10−5 mbar), the amount of isolated silanols (measured by titration with MeLi)53 is about 1.8 mmol g−1 (1.3 OH nm−2).The reaction of 1 equivalent of TIBA (1 M/hexane)/silanol with SBA15700 leads to the formation of several compounds: [(SiO)2–Al–CH2CH(CH3)2] 1a, silicon isobutyl [Si–CH2CH(CH3)2] 1b and silicon hydride [Si–H] 1c (Scheme 2). These results indicate that TIBA reacts not only with single silanols (Scheme 2, pathway a) but also with strained siloxane bridges (Scheme 2, pathway b).41,54 Interestingly – the amount of gas phase isobutane plus isobutene is close to the amount of grafted Al in principle in agreement with a protonolysis of one Al-isobutyl group and the microanalysis gives 8C/grafted Al (Tables S1 and S2, ESI†) – surprisingly the hydrolysis of the material gives only one mole of isobutane per grafted Al indicating that a Si-isobutyl, which is not hydrolyzed, is already formed at room temperature simultaneously to Al-isobutyl.
The IR spectra obtained after reaction with TIBA (1 eq./SiOH) are shown in Fig. 1.
 | ||
| Fig. 1 IR spectra of (i) SBA15700 (blue), (ii) SBA15700 after reaction with 1 equiv. of TIBA/SiOH (red) and (ii) − (i) subtraction of spectra (ii) and (i) (black). | ||
As expected, a partial consumption of the Si–OH vibrational band is observed [3741 cm−1, ν(OH)]. Vibrational isobutyl groups appear at 2950 [νas(CH3)], 2865 [νs(CH2)], 1465 [δas(CH3)] and 1365 cm−1 [δs(CH3)]. Reaction of TIBA also led to the formation of silicon hydride(s) species characterized by a broad ν(Si–H) band at 2100–2200 cm−1.39,55,56 Based on DFT calculations (vide infra), the presence of these silicon hydrides is explained by a β-H elimination from one of the isobutyl fragment to the Al center of [SiO–Al–[CH2CH(CH3)2]2. Then, the Al–H intermediate thus formed reacts with adjacent silicon bridges to form silicon hydride.
The proton solid-state NMR spectrum of 1 displays five clear signals at 0.4, 0.9, 1.7, 2 and 4 ppm (Fig. S1, ESI†). According to both literature data and liquid state NMR of TIBA (Fig. S2 and S3, ESI†), the signals at 0.4, 0.9 and 2 ppm are assigned respectively to [(SiO)2–Al–C![[H with combining low line]](https://www.rsc.org/images/entities/b_i_char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/b_char_0032_0332.gif) CH(CH3)2], [(SiO)2–Al–CH2CH(C
CH(CH3)2], [(SiO)2–Al–CH2CH(C![[3 with combining low line]](https://www.rsc.org/images/entities/b_char_0033_0332.gif) )2] and [(SiO)2–Al–CH2C(CH3)2]. The silicon hydride proton chemical shift appears at 4 ppm55,57 as expected from the FT-IR results.
)2] and [(SiO)2–Al–CH2C(CH3)2]. The silicon hydride proton chemical shift appears at 4 ppm55,57 as expected from the FT-IR results.
Finally, the shoulder at 0.8 ppm is attributed to silicon isobutyl moiety [Si–CCH(C)2] which was suggested to be formed from analytical data.58,59 Another peak at 1.7 ppm is ascribed to unreacted silanols.
The 2D double quantum (DQ) 1H-1H MAS NMR spectrum shows for the isobutyl group [CCH(C)]2 an intense autocorrelation on the 2:1 diagonal centered around 0.4–0.9 ppm in F2 and 0.8–1.8 ppm. Outside the diagonal, a strong correlation is observed at 2 ppm assigned to [Si–CH2C(CH3)2] and [(SiO)2–Al–CH2C(CH3)2] (Fig. 2i).
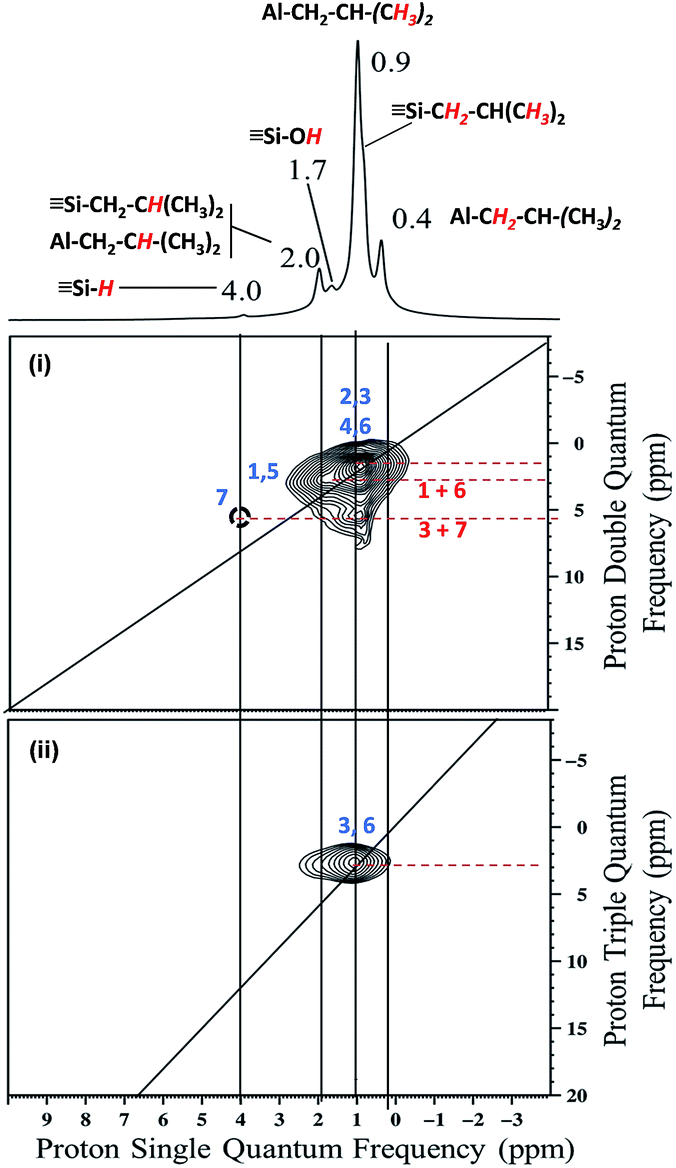 | ||
| Fig. 2 (i) DQ rotor-synchronized 2D 1H MAS NMR spectrum of 1. (ii) 1H TQ MAS spectra of 1. | ||
This means that in the isobutyl fragments the 1H on the αC and βC are close together (Scheme 3a).
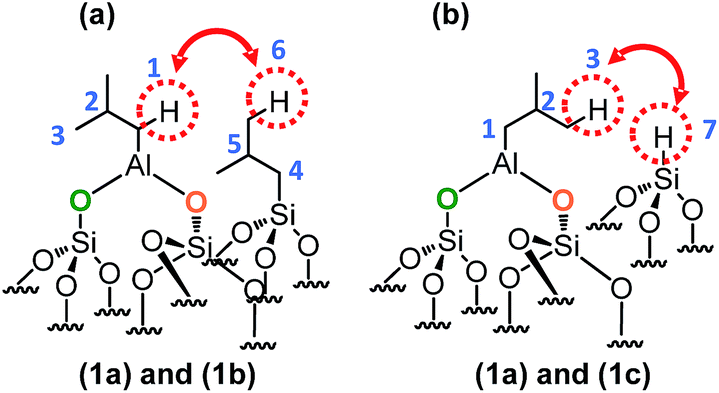 | ||
| Scheme 3 Representation of the close proximity of (a) aluminum isobutyl 1a and silicon isobutyl 1b, (b) aluminum isobutyl 1a and silicon hydride 1c. | ||
The proton resonance assigned to [Si–] at 4 ppm shows a strong correlation with the proton at 0.9 ppm assigned to the methyl group (Fig. 2i). In other words, [Si–H] and [(SiO)2–Al–CH2CH(CH3)2] species are in close proximity (<5 Å) (Scheme 3b).
This suggests strongly that they were formed from the same elementary step on the surface.
The 1H triple quantum (TQ) solid state NMR spectrum of 1 highlights the expected correlation at 2.7 ppm corresponding to the proton of methyl group of [(SiO)2–Al–CH2CH(C)2] and [Si–CH2CH(CH3)2] species, [3*δH(CH3) = 3*0.9] (Fig. 2ii).
13C cross-polarization magic angle spinning (CP-MAS) spectrum shows a broad signal center at 24 ppm, assigned to Al–![[C with combining low line]](https://www.rsc.org/images/entities/b_i_char_0043_0332.gif) H2–CH–(CH3)2 and Si–CH2–H–(H3)2 associated with two shoulders at 23 and 26 ppm attributed to Si–H2– and Al–CH2–H–(H3)2, respectively (Fig. S4, ESI†).41 The 29Si NMR spectrum of 1 exhibits two signals centered at −106 ppm (broad) and −47 ppm (weak) (Fig. S5, ESI†). According to the literature, the former is attributed to both [(SiO)3SiOE], with E = Si or H (commonly called Q4 and Q3) and Si(OAl) (OSi)360 and the latter is assigned to [(SiO)2Si(OH)X], with X = H or R (commonly called T2 site).55 Interestingly, the 1:1 ratio between TIBA/SiOH used in this work induces no significant Al incorporation in network as compared to Coperet et al. works.42 Indeed, no signal characteristic of Si(OAl)4 at – 85 ppm is observed.60
H2–CH–(CH3)2 and Si–CH2–H–(H3)2 associated with two shoulders at 23 and 26 ppm attributed to Si–H2– and Al–CH2–H–(H3)2, respectively (Fig. S4, ESI†).41 The 29Si NMR spectrum of 1 exhibits two signals centered at −106 ppm (broad) and −47 ppm (weak) (Fig. S5, ESI†). According to the literature, the former is attributed to both [(SiO)3SiOE], with E = Si or H (commonly called Q4 and Q3) and Si(OAl) (OSi)360 and the latter is assigned to [(SiO)2Si(OH)X], with X = H or R (commonly called T2 site).55 Interestingly, the 1:1 ratio between TIBA/SiOH used in this work induces no significant Al incorporation in network as compared to Coperet et al. works.42 Indeed, no signal characteristic of Si(OAl)4 at – 85 ppm is observed.60
Finally, we performed 27Al NMR spectroscopy. This technique is usually an efficient probe to demonstrate the presence of alkyl group and to assess the coordinance of Al-alkyl center.40,61 The 27Al NMR spectrum of material 1 shows three clear signals at 5, 33 and 59 ppm (Fig. S6, ESI†) typically assigned to octahedral (AlOh), pentahedral (AlPh) and tetrahedral (AlTh),62–65 but not characteristic of alkyl complexes. Indeed, a tetrahedral Al-alkyl features a chemical shift of 117 ppm and a quadrupolar coupling constant of 17.5 MHz.66 However, as previously describes by Espinas et al.27Al NMR signals are notoriously difficult to observe to very broad line shape.41 As frequently observed in SOMC,2 Al center coordinate to the silica surface (Si–O–Si) affording Al-surfaces complexes with different Al chemical shifts. The AlOh species are closer to the surface and so are assumed to be due to the reaction of TIBA with the strained siloxane whereas the AlTh species are issued from the reaction of single silanol with TIBA. AlPh is probably the result of both reactions. Thus, material 1 displays three types of isobutyl aluminum geometries along isobutyl silicon and silicon hydride. To have a more detailed understanding of the reactivity of TIBA with SBA15700 a mechanistic rationalization of these results will be provided in the following section.
Density functional theory calculations
The experimental results described so far can be explained with a variety of mechanisms. To achieve a more structured understanding of these systems we performed DFT calculations. We first report on the reactivity of TIBA with a siloxane bridge on the silica surface, then we move to analyze the reactivity of TIBA with a silanol protruding from the silica surface, and we continue with conversion of monopodal [SiO–Al–[CH2CH(CH3)2]2 into bipodal [(SiO)2–Al–CH2CH(CH3)2].
Reaction of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) Si–O–Si with TIBA
Si–O–Si with TIBA
The initial step is coordination of the oxophilic Al center of TIBA to an O atom on the silica surface, to give intermediate a1 with release of 7.0 kcal mol−1 (Fig. 3). For the sake of simplicity, in the following we will assume intermediate a1 as reference structure at 0 kcal mol−1 in energy. Reactivity of a1 can involve either the transfer of one of the isobutyl groups of TIBA to a Si atom, or the transfer of a β-H atom from one of the isobutyl groups of TIBA to a Si atom, with release of isobutene. The alkyl transfer pathway (blue profile) occurs through the four centers transition state a1-a2, 16.2 kcal mol−1 above a1. Product a2, presenting the [SiO–Al–[CH2CH(CH3)2]2] and [Si–CH2CH(CH3)2] moieties, lies 14.5 kcal mol−1 below the initial adduct a1. The β-H transfer pathway (red profile) occurs through the six centers transition state a1-a2′, 42.2 kcal mol−1 above a1, and collapses into the Al-coordinated isobutene intermediate a2′, 7.6 kcal mol−1 above a1.
 | ||
| Fig. 3 Energy profile for the reactivity of TIBA with siloxane bridges (Si–O–Si). | ||
Finally, isobutene dissociation leads to species a3′. Comparison of the two pathways of Fig. 3 clearly indicates that the only viable mechanism corresponds to alkyl transfer to form a2.
At this point, we should model reactivity of the formed monopodal [SiO–Al–[CH2CH(CH3)2]2] with a nearby siloxane bridge to yield bipodal [(SiO)2–Al–CH2CH(CH3)2]. However, since this conversion can also occur with a monopodal Al species deriving from initial reactivity of TIBA with a silanol (Scheme 2), we decided to use a general model for the monopodal Al to bipodal Al conversion, see the next section.
Reaction of Si–OH with TIBA
The first step is the coordination of TIBA to the silanol, resulting in the intermediate b1, with an energy gain of 10.3 kcal mol−1 (Fig. 4).
 | ||
| Fig. 4 Energy profile for the reactivity of TIBA with isolated silanol (Si–OH). | ||
The reaction occurs via σ-bond metathesis with release of isobutane through the four centers transition state b1-b2, 6.2 kcal mol−1 above b1. This leads to the monopodal [SiO–Al–[CH2CH(CH3)2]2] b2, 49.5 kcal mol−1 below b1.
As the starting point of the second step corresponds to the monopodal structure, in the following, we will assume intermediate b2 as reference structure at 0 kcal mol−1 in energy. We envisaged three possible evolutions for intermediate b2. The first corresponds to the transfer of an isobutyl group from b2 to a siloxane bridge on the surface, with formation of bipodal [(SiO)2–Al–CH2CH(CH3)2] and [Si–CH2CH(CH3)2], b3 (Fig. 5, Scheme S1, ESI†). The second corresponds to the transfer of a β-H atom from one isobutyl group of b2 to a siloxane bridge, with formation of bipodal [(SiO)2–Al–CH2CH(CH3)2] and of silicon hydride [Si–H], b5′′ associated with the release of isobutene (Fig. 5, Scheme S2, ESI†). The third pathway starts with a β-H elimination from one of the isobutyl to the Al center, followed by the transfer of this H atom from Al to a siloxane bridge.
 | ||
| Fig. 5 Energy profile for the reactivity of monopodal [SiO–Al–[CH2CH(CH3)2]2] with siloxane bridges, (Si–O–Si). Alkyl transfer versus direct H transfer. | ||
Focusing on the first pathway, the alkyl transfers occurs through the four centers transition state b2-b3, with a barrier of 24.6 kcal mol−1 from b2 (Fig. 5, blue profile).
Moving to the second pathway, the β-H transfer to the Si with elimination of isobutene occurs through the six centers transition state b2-b3′, with a barrier of 46.7 kcal mol−1 from b2. This result excludes this pathway from a possible reaction scenario (Fig. 5, red profile). The third pathway starts with a β-H elimination from one of the isobutyl to the Al center via transition state b2-b3′′, 25.6 kcal mol−1 above b2, and formation of intermediate b3′′. This intermediate presents an Al–H bond and an isobutene molecule still coordinated to the Al center. The dissociation of isobutene leads to intermediate b4′′, which presents an interaction between the Al center and an O atom of a nearby siloxane bridge. The transfer of the H center to the Si atom of the siloxane bridge occurs through the four centers transition state b4′′-b5′′, with a barrier of 10.4 kcal mol−1 from b4′′. The final product b5′′ presents the bipodal [(SiO)2–Al–CH2CH(CH3)2] and [Si–H] (Fig. 6, green profile and Scheme S3, ESI†).
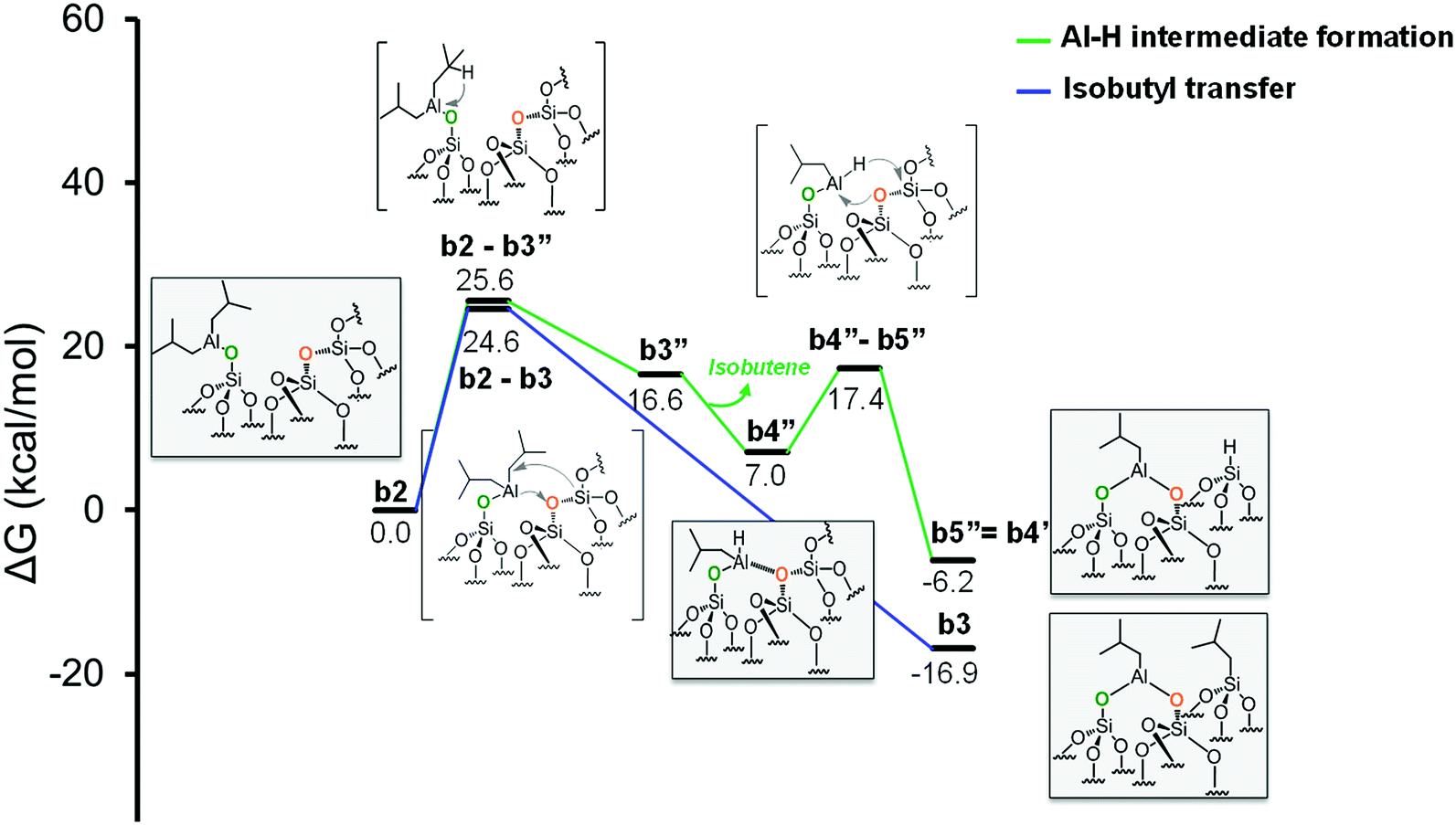 | ||
| Fig. 6 Energy profile for the reactivity of monopodal [SiO–Al–[CH2CH(CH3)2]2] with siloxane bridges, (Si–O–Si). Alkyl transfer versus two step H transfer. | ||
While our results should be taken cum grano salis, since the exact energetics of the various transformations certainly depends on the specific model we used for the underlying silica surface, we believe they allow depicting a well-grounded scenario. First, alkyl transfer from b2 to a siloxane bridge is clearly favored over β-H transfer with isobutene release, both kinetically and thermodynamically. Secondly, the barrier for the alkyl transfer to a siloxane bridge is of moderately low energy even with the poorly strained silica models we used. Third, reactivity of TIBA with the silanol is easier than reactivity of TIBA with a siloxane bridge, which is in agreement with the experiments discussed above. Fourth, formation of Si–H bonds can be only explained by a two-step mechanism, with initial β-H elimination to the Al, and formation of an Al–H bond (Fig. 7a), followed by H-transfer to a siloxane bridge. This reaction is in competition with the alkyl-transfer, selectivity being determined at the level of transition states b2-b3 and b2-b3′′. With the specific model for silica we used, the alkyl transfer transition state b2-b3 is favored by only 1 kcal mol−1 over the β-H elimination transition state b2-b3′′. Decomposition of this small free energy difference into electronic and thermal (including zero-point energy, ZPE, correction) contributions indicate that transition state b2-b3 is favored by 9.5 kcal mol−1 in terms of electronic energy, and it is the thermal plus ZPE terms that reduce remarkably the free energy gap between the two transition states. This effect can be rationalized considering that transition state b2-b3 is highly ordered, with the Al atoms strongly engaged with a siloxane bridge, which reduces remarkably the flexibility of the Al moiety, (Fig. 7b). Conversely, the β-H elimination transition state b2-b3′′ is much more flexible, since the elimination occurs within the Al moiety, without any interaction with the silica surface, (Fig. 7c). This is well indicated by the presence of three vibrational modes of very low frequency (below 20 cm−1) in transition state b2-b3′′, corresponds to librational movements of the Al moiety. Despite the scarce accuracy of these low frequency vibrational modes within the harmonic approximation, which affects the exact value of the free energy gap between transition states b2-b3 and b2-b3′′, they clearly provide an explanation for the competition between alkyl and β-H transfer to the silica surface.
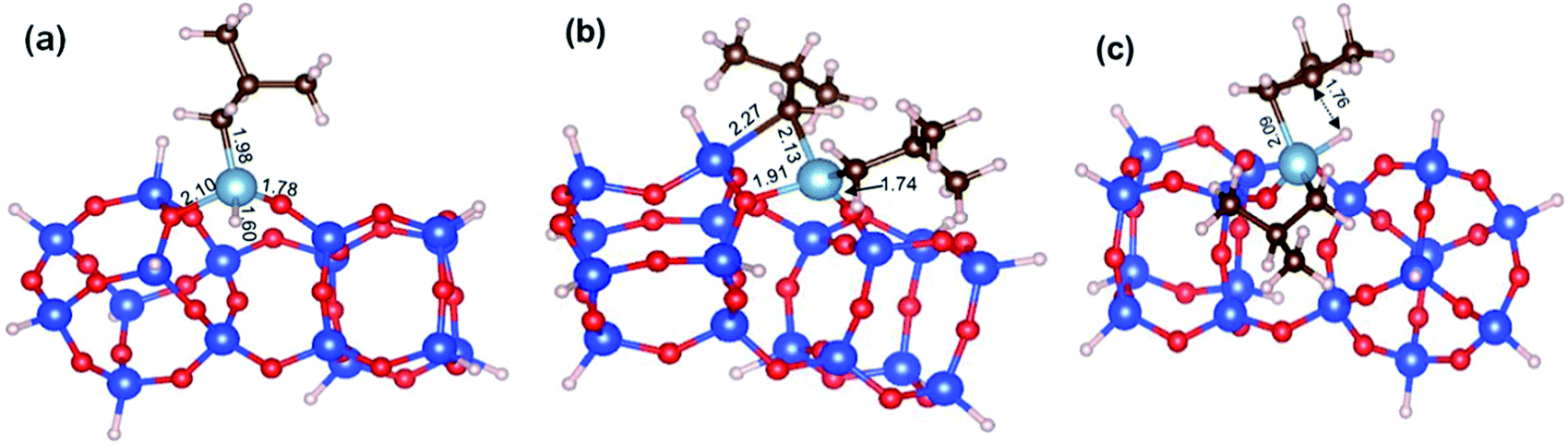 | ||
| Fig. 7 DFT optimized geometry of (a) the Al–H intermediate b3′′, (b) of the transition states b2-b3 and b2-b3′′, for alkyl transfer to the silica, and (c) the β-H elimination to the Al. Distances are in Å. | ||
Furthermore, DFT calculations are in qualitative agreement with the experimental data obtained from elemental and gas phase analysis. Indeed, quantification of both processes: alkyl transfer vs. two step H transfer (Fig. 6) is experimentally possible. The total amount of Al atoms in the surface is 1.35 mmol g−1 (Table S1†). The amount of isobutene, coming from the generation of Si–H via Al–H intermediate and due to the two step H transfer, is 0.5 mmol g−1 (Table S2†). This two step H transfer process corresponds to the ratio between the amount of isobutene and the amount of total Al. Finally, the two step H transfer occurs at 37% and the alkyl transfer at 63%. The latter is favored according to both experimental and DFT calculations.
Generation of aluminum hydrides species
Aluminum hydride have been already formed on γ-alumina but the process required harsh temperature conditions (400 °C) under H2 pressure of 0.733 bar.43 We were interested in the formation of aluminum hydride species through a variable temperature treatment (100 °C to 400 °C, rate 60 °C h−1, Fig. S8 and S9, ESI†) under high vacuum (10−5 mbar). This process was followed by FT-IR spectroscopy and the best temperature to achieve the formation of aluminum hydride is 225 °C (Fig. S9 and S10,†Scheme 4). | ||
| Scheme 4 Formation of aluminum hydride through a thermal treatment (100 °C to 400 °C) under 10−5 mbar for 20 h of 1. | ||
At 225 °C, the FT-IR spectrum shows a strong decrease of the vibrational bands of isobutyl groups at 2950 [νas(CH3)], 2865 [νs(CH2)], 1465 [δas(CH3)] and 1365 cm−1 [δs(CH3)].
Besides, three new bands appear at 1940, 1664 and 1610 cm−1 (Fig. 8).43,67 They are easily attributed to the vibration bands of Al–H. Indeed, terminal Al–H elongation bands range down to 1800 cm−1, bridging Al–H–Al give rise to signals between 1750 and 1550 cm−1.67 Thus, the β-H elimination occurs from Al-isobutyl and results on the formation of Al–H (terminal and bridging) characterized by vibration bands at 1940 and 1664, 1610 cm−1, respectively. Note, that a hydride transfer from Al–H to closer siloxane could also occur with the temperature to form either silicon hydride or bis-hydride characterized by two ν(Si–H) band at 2250 and 2156 cm−1, respectively. At 300 °C, the vibration band assigned to Al–H decreases significantly upon the increase of the characteristic vibration band of silicon hydride. Above 300 °C, no Al–H remains, only the silicon monohydride species are present.68 But it is interesting to mention that the aluminum hydride species formed at 225 °C are highly stable with time at room temperature under static vacuum (Fig. S10, ESI†).
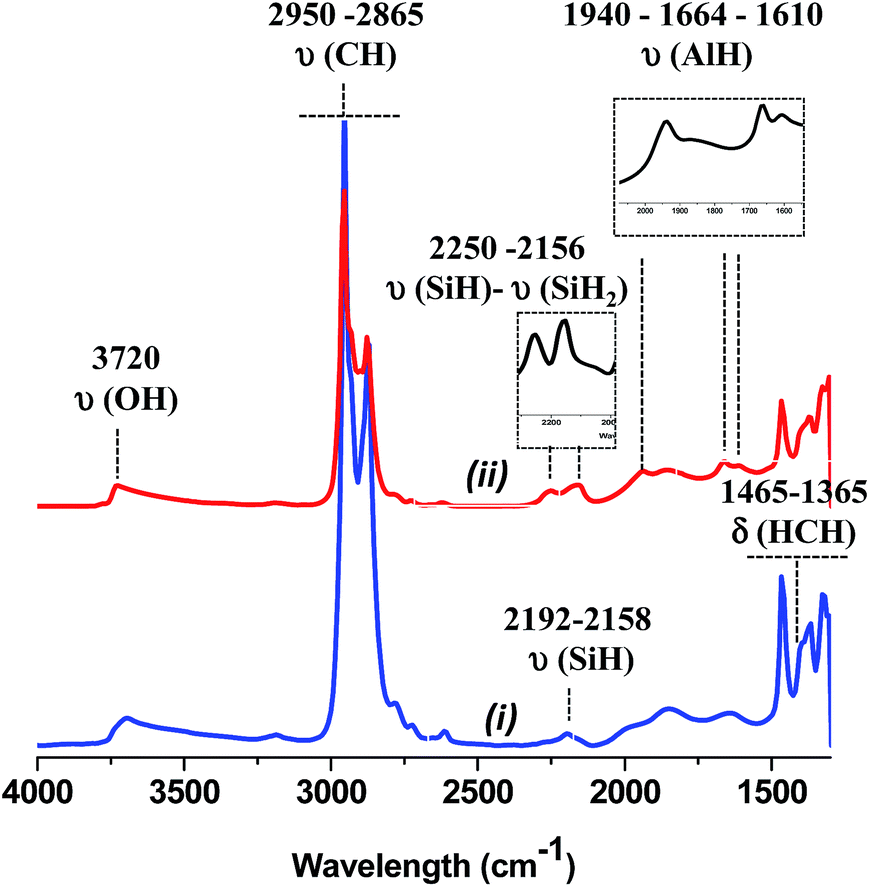 | ||
| Fig. 8 (i) IR spectra of 1, (ii) after thermal treatment at 225 °C for 24 h. Inlet subtraction (ii) − (i) in the corresponding area of silicon and aluminum hydride, respectively. | ||
To reinforce these results we performed solid state NMR spectroscopies of sample treated at 225 °C for 24 h under dynamic vacuum (Fig. 9).
 | ||
| Fig. 9 (i) DQ rotor-synchronized 2D 1H MAS NMR spectrum of 2. (ii) 1H TQ MAS spectra of 2 (7′ is ascribed for SiH2). | ||
The 1H NMR spectrum of 2 shows the disappearance of the signal characteristic of [(SiO)2–Al–CHCH(CH3)2] species at 0.4 ppm. Only the silicon isobutyl remains on the surface, as the peaks at 0.9 and 2 ppm are still present. It is in accordance with the 13C CP-MAS NMR spectrum where only the chemical shift at 24 ppm is observed (Fig. S11, ESI†). The signal at 26 ppm assigned to Al–CH2–CH–(CH3)2 is no longer detected. The broad 1H signal at 4.3 ppm is attributed to Al–H,43 Si–H and SiH.55,56,68
In the 2D 1H-1H and in TQ spectra (Fig. 9), a strong auto-correlation is observed at 0.9 ppm. It corresponds to the proton signal of the [Si–C![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) –CH (C
–CH (C![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) )2]. Besides in the spectrum of Fig. 9i, a strong correlation is observed between the proton of Al–H and the [Si–CH2–CH(CH)2]. In other words, Al–H and silicon isobutyl are in close proximity (<5 Å) (Scheme 5a).
)2]. Besides in the spectrum of Fig. 9i, a strong correlation is observed between the proton of Al–H and the [Si–CH2–CH(CH)2]. In other words, Al–H and silicon isobutyl are in close proximity (<5 Å) (Scheme 5a).
 | ||
| Scheme 5 Representation of the close proximity of (a) aluminum hydride 2a and silicon isobutyl 2b, (b) aluminum hydride 2a and silicon hydride 2c. | ||
Besides, Al–H and Si–H are also close (Scheme 5b) due to the correlation centered at 8.6 ppm [δH(Al–H) + [δH(Si–H) = 4.3 + 4.3]. Finally to confirm the presence of the different coordination mode of the Al–H species, 27Al solid state NMR was performed (Fig. 10a). As expected and in accordance with the FT-IR studies, AlTh/Ph hydride species are assumed to be present in more important proportion than AlOh hydride species. The latter one reacts with closer siloxane bridge to form silicon hydride species. The 27Al–1H correlation spectrum in Fig. 10b was acquired using the J-HMQC pulse sequence which correlates nuclei through their scalar coupling, a method well suited for hydride species presenting large 1JAl–H couplings. The broad signal along the 27Al dimension corresponds to only those aluminum centers bearing hydride ligands. Thus, the proton signal at 4.3 ppm correlates with the 27Al signals at 3, 31 and 51 ppm corresponding respectively to octa, penta and tetrahedral aluminum hydride.
 | ||
| Fig. 10 (a) 27Al MAS solid-state NMR spectrum of 2 and (b) 27Al–1H J-HMQC MAS NMR spectrum of 2. | ||
The spectrum of Fig. 10b clearly shows that AlOh are more abundant than AlTh/Ph. Interestingly, the spectrum of Fig. 10a, reveals the opposite: few AlOh at 3 ppm. Thus, the projection of the 2D 1H–27Al J-HMQC MAS NMR spectrum in the 27Al dimension was performed to provide some semi-quantitative information regarding the coordinance of supported Al–H (Fig. S12†). These data confirm the predominance of AlTh/Ph towards AlOh. Finally, the J-HMQC-filtered 1H MAS spectrum (Fig. S13†) shows the presence of a 1H site at a chemical shift of 4 ppm, in a major proportion. This chemical shift lies above the expected range for molecular aluminum hydrides.69
Conclusions
The mechanism of reaction of TIBA with SBA15700 is clearly elucidated by combining experimental data and DFT calculations. Both show the formation of a monopodal bis-isobutyl aluminum intermediate due to simultaneous protonolysis and opening siloxane bridge. This intermediate leads to a bipodal bissiloxy isobutyl aluminum involving the formation of either a silicon isobutyl (alkyl transfer) or a silicon hydride (β-H elimination via aluminum hydride intermediate). From this material 1, we are able to generate aluminum hydride 2 through a thermal treatment at 225 °C under high vacuum. This is possible by a β-H elimination. All these supported aluminum materials were fully characterized by FT-IR and advanced solid state NMR spectroscopies. The results are in accordance with the formation of tetra, penta and octahedral stable aluminum hydride. This study is a key step for the design of single site silica supported aluminum (alkyl or hydride).Acknowledgements
We gratefully acknowledge the King Abdullah University of Science and Technology for financial support.Notes and references
- R. A. van Santen and M. Neurock, Molecular Heterogeneous Catalysis, 2009 Search PubMed.
- J. M. Basset, R. Psaro, D. Roberto and R. Ugo, Modern Surface Organometallic Chemistry, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed.
- E. Lucenti, D. Roberto and R. Ugo, Organometallics, 2001, 20, 1725–1733 CrossRef CAS.
- J. M. Basset, S. L. Scott, A. Choplin, M. Leconte, F. Quignard, C. Santini and A. Theolier, in Elementary Reaction Steps in Heterogeneous Catalysis, ed. R. W. Joyner and R. A. VanSanten, 1993, vol. 398, pp. 39–49 Search PubMed.
- J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073–4147 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- W. Gu, M. M. Stalzer, C. P. Nicholas, A. Bhattacharyya, A. Motta, J. R. Gallagher, G. Zhang, J. T. Miller, T. Kobayashi, M. Pruski, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 6770–6780 CrossRef CAS PubMed.
- R. Schowner, W. Frey and M. R. Buchmeiser, J. Am. Chem. Soc., 2015, 137, 6188–6191 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- M. Stalzer, M. Delferro and T. Marks, Catal. Lett., 2015, 145, 3–14 CrossRef CAS.
- C. Deraedt, M. d'Halluin and D. Astruc, Eur. J. Inorg. Chem., 2013, 2013, 4881–4908 CAS.
- M. R. Buchmeiser, Chem. Rev., 2009, 109, 303–321 CrossRef CAS PubMed.
- P. Serna and B. C. Gates, Acc. Chem. Res., 2014, 47, 2612–2620 CrossRef CAS PubMed.
- K. M. Engle, G. Lu, S.-X. Luo, L. M. Henling, M. K. Takase, P. Liu, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2015, 137, 5782–5792 CrossRef CAS PubMed.
- B. Hamzaoui, M. E. Eter, E. Abou-hamad, Y. Chen, J. D. A. Pelletier and J.-M. Basset, Chem.–Eur. J., 2015, 21, 4294–4299 CrossRef CAS PubMed.
- A. Hamieh, Y. Chen, S. Abdel-Azeim, E. Abou-hamad, S. Goh, M. Samantaray, R. Dey, L. Cavallo and J. M. Basset, ACS Catal., 2015, 5, 2164–2171 CrossRef CAS.
- B. A. Morrow, in Stud. Surf. Sci. Catal., ed. J. L. G. Fierro, Elsevier, 1990, vol. 57, Part A, pp. A161–A224 Search PubMed.
- E. F. Vansant, P. van Der Voort and K. C. Vrancken, in Studies in Surface Science and Catalysis, Elsevier, 1995, vol. 93 Search PubMed.
- N. Merle, J. Trébosc, A. Baudouin, I. D. Rosal, L. Maron, K. Szeto, M. Genelot, A. Mortreux, M. Taoufik, L. Delevoye and R. M. Gauvin, J. Am. Chem. Soc., 2012, 134, 9263–9275 CrossRef CAS PubMed.
- M. K. Samantaray, R. Dey, E. Abou-Hamad, A. Hamieh and J.-M. Basset, Chem.–Eur. J., 2015, 21, 6100–6106 CrossRef CAS PubMed.
- B. A. Morrow and I. A. Cody, J. Phys. Chem., 1976, 80, 1995–1998 CrossRef CAS.
- K. Amakawa, L. Sun, C. Guo, M. Hävecker, P. Kube, I. E. Wachs, S. Lwin, A. I. Frenkel, A. Patlolla, K. Hermann, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2013, 52, 13553–13557 CrossRef CAS PubMed.
- L. H. Dubois and B. R. Zegarski, J. Am. Chem. Soc., 1993, 115, 1190–1191 CrossRef CAS.
- A. Grabbe, T. A. Michalske and W. L. Smith, J. Phys. Chem., 1995, 99, 4648–4654 CrossRef CAS.
- S. D. Fleischman and S. L. Scott, J. Am. Chem. Soc., 2011, 133, 4847–4855 CrossRef CAS PubMed.
- W. M. H. Sachtler, Catal. Rev., 1976, 14, 193–210 CAS.
- W. M. H. Sachtler, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knozinger and J. Weitkamp, Wiley-VCH, Weinheim, 1997, vol. 3, pp. 1077–1084 Search PubMed.
- W. M. H. Sachtler, Vide, 1973, 164, 67 Search PubMed.
- R. Anwander, C. Palm, O. Groeger and G. Engelhardt, Organometallics, 1998, 17, 2027–2036 CrossRef CAS.
- J. Li, J. A. DiVerdi and G. E. Maciel, J. Am. Chem. Soc., 2006, 128, 17093–17101 CrossRef CAS PubMed.
- M. J. D. Low, A. G. Severdia and J. Chan, J. Catal., 1981, 69, 384–391 CrossRef CAS.
- M. D. Skowronska-Ptasinska, R. Duchateau, R. A. van Santen and G. P. A. Yap, Organometallics, 2001, 20, 3519–3530 CrossRef CAS.
- D. J. C. Yates, G. W. Dembinski, W. R. Kroll and J. J. Elliott, J. Phys. Chem., 1969, 73, 911–921 CrossRef CAS.
- M. E. Bartram, T. A. Michalske and J. W. Rogers, J. Phys. Chem., 1991, 95, 4453–4463 CrossRef CAS.
- R. N. Kerber, A. Kermagoret, E. Callens, P. Florian, D. Massiot, A. Lesage, C. Copéret, F. Delbecq, X. Rozanska and P. Sautet, J. Am. Chem. Soc., 2012, 134, 6767–6775 CrossRef CAS PubMed.
- S. Ewart and M. Baird, Top. Catal., 1999, 7, 1–8 CrossRef CAS.
- K.-T. Li and Y.-T. Kao, J. Appl. Polym. Sci., 2006, 101, 2573–2580 CrossRef CAS PubMed.
- J. B. Kinney and R. H. Staley, J. Phys. Chem., 1983, 87, 3735–3740 CrossRef CAS.
- S. Charoenchaidet, S. Chavadej and E. Gulari, Macromol. Rapid Commun., 2002, 23, 426–431 CrossRef CAS.
- R. Benn, E. Janssen, H. Lehmkuhl and A. Rufínska, J. Organomet. Chem., 1987, 333, 155–168 CrossRef CAS.
- J. Pelletier, J. Espinas, N. Vu, S. Norsic, A. Baudouin, L. Delevoye, J. Trebosc, E. Le Roux, C. Santini, J.-M. Basset, R. M. Gauvin and M. Taoufik, Chem. Commun., 2011, 47, 2979–2981 RSC.
- A. Kermagoret, R. N. Kerber, M. P. Conley, E. Callens, P. Florian, D. Massiot, C. Coperet, F. Delbecq, X. Rozanska and P. Sautet, Dalton Trans., 2013, 42, 12681–12687 RSC.
- E. Mazoyer, J. Trébosc, A. Baudouin, O. Boyron, J. Pelletier, J.-M. Basset, M. J. Vitorino, C. P. Nicholas, R. M. Gauvin, M. Taoufik and L. Delevoye, Angew. Chem., Int. Ed., 2010, 49, 9854–9858 CrossRef CAS PubMed.
- M. G. Klimpel, J. Eppinger, P. Sirsch, W. Scherer and R. Anwander, Organometallics, 2002, 21, 4021–4023 CrossRef CAS.
- G. W. Coates, P. D. Hustad and S. Reinartz, Angew. Chem., Int. Ed., 2002, 41, 2236–2257 CrossRef CAS.
- H. Sinn and W. Kaminsky, in Adv. Organomet. Chem., ed. F. G. A. Stone and W. Robert, Academic Press, 1980, vol. 18, pp. 99–149 Search PubMed.
- A. Tarazona, E. Koglin, F. Buda, B. B. Coussens, J. Renkema, S. van Heel and R. J. Meier, J. Phys. Chem. B, 1997, 101, 4370–4378 CrossRef CAS.
- K. Soga and T. Shiono, Prog. Polym. Sci., 1997, 22, 1503–1546 CrossRef CAS.
- A. Fischbach, F. Perdih, P. Sirsch, W. Scherer and R. Anwander, Organometallics, 2002, 21, 4569–4571 CrossRef CAS.
- L. R. Rieth, R. F. Eaton and G. W. Coates, Angew. Chem., Int. Ed., 2001, 40, 2153–2156 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- S. C. Antakli and J. Serpinet, Chromatographia, 1987, 23, 767–769 CAS.
- S. L. Scott and J.-M. Basset, J. Am. Chem. Soc., 1994, 116, 12069–12070 CrossRef CAS.
- F. Rataboul, A. Baudouin, C. Thieuleux, L. Veyre, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2004, 126, 12541–12550 CrossRef CAS PubMed.
- A. Bendjeriou-Sedjerari, J. M. Azzi, E. Abou-Hamad, D. H. Anjum, F. A. Pasha, K.-W. Huang, L. Emsley and J.-M. Basset, J. Am. Chem. Soc., 2013, 135, 17943–17951 CrossRef CAS PubMed.
- H. J. Campbell-Ferguson, E. A. V. Ebsworth, A. G. MacDiarmid and T. Yoshioka, J. Phys. Chem., 1967, 71, 723–726 CrossRef CAS.
- S. E. Anderson, A. Somogyi, T. S. Haddad, E. B. Coughlin, G. Gadodia, D. F. Marten, J. Ray and M. T. Bowers, Int. J. Mass Spectrom., 2010, 292, 38–47 CrossRef CAS PubMed.
- S.-C. Chan, S.-W. Kuo, H.-S. She, H.-M. Lin, H.-F. Lee and F.-C. Chang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 125–135 CrossRef CAS PubMed.
- E. Lippmaa, M. Maegi, A. Samoson, M. Tarmak and G. Engelhardt, J. Am. Chem. Soc., 1981, 103, 4992–4996 CrossRef CAS.
- R. Benn, A. Rufínska, H. Lehmkuhl, E. Janssen and C. Kruger, Angew. Chem., Int. Ed. Engl., 1983, 22, 779–780 CrossRef PubMed.
- A. Goldbourt, M. V. Landau and S. Vega, J. Phys. Chem. B, 2002, 107, 724–731 CrossRef.
- L. A. O'Dell, S. L. P. Savin, A. V. Chadwick and M. E. Smith, Solid State Nucl. Magn. Reson., 2007, 31, 169–173 CrossRef PubMed.
- V. F. F. Barbosa, K. J. D. MacKenzie and C. Thaumaturgo, Int. J. Inorg. Mater., 2000, 2, 309–317 CrossRef CAS.
- M. Baca, E. de la Rochefoucauld, E. Ambroise, J.-M. Krafft, R. Hajjar, P. P. Man, X. Carrier and J. Blanchard, Microporous Mesoporous Mater., 2008, 110, 232–241 CrossRef CAS PubMed.
- F.-J. Wu, L. S. Simeral, A. A. Mrse, J. L. Eilertsen, L. Negureanu, Z. Gan, F. R. Fronczek, R. W. Hall and L. G. Butler, Inorg. Chem., 2007, 46, 44–47 CrossRef CAS PubMed.
- M. Veith, J. Frères, V. Huch and M. Zimmer, Organometallics, 2006, 25, 1875–1880 CrossRef CAS.
- G. Tosin, M. Delgado, A. Baudouin, C. C. Santini, F. Bayard and J.-M. Basset, Organometallics, 2010, 29, 1312–1322 CrossRef CAS.
- D. Healy, M. R. Mason, P. W. Gravelle, S. G. Bott and A. R. Barron, J. Chem. Soc., Dalton Trans., 1993, 441–454 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02276b |
| This journal is © The Royal Society of Chemistry 2015 |